使用 Pico488 进行 DNA 定量
Pico488 DNA 定量溶液是一种超灵敏试剂,用于在无法通过测量 260 nm 吸光度来确定双链 DNA 浓度时测量双链 DNA 浓度。 Pico488 选择性地结合双链 DNA,因此核苷酸、单链 DNA、RNA、蛋白质和其他杂质不会妨碍测量。与双链 DNA 结合的染料在 503 nm 处最大吸收光并在 525 nm 处最大发射光。为了检测测定读数,可以使用任何类型的荧光计或荧光板读数器。
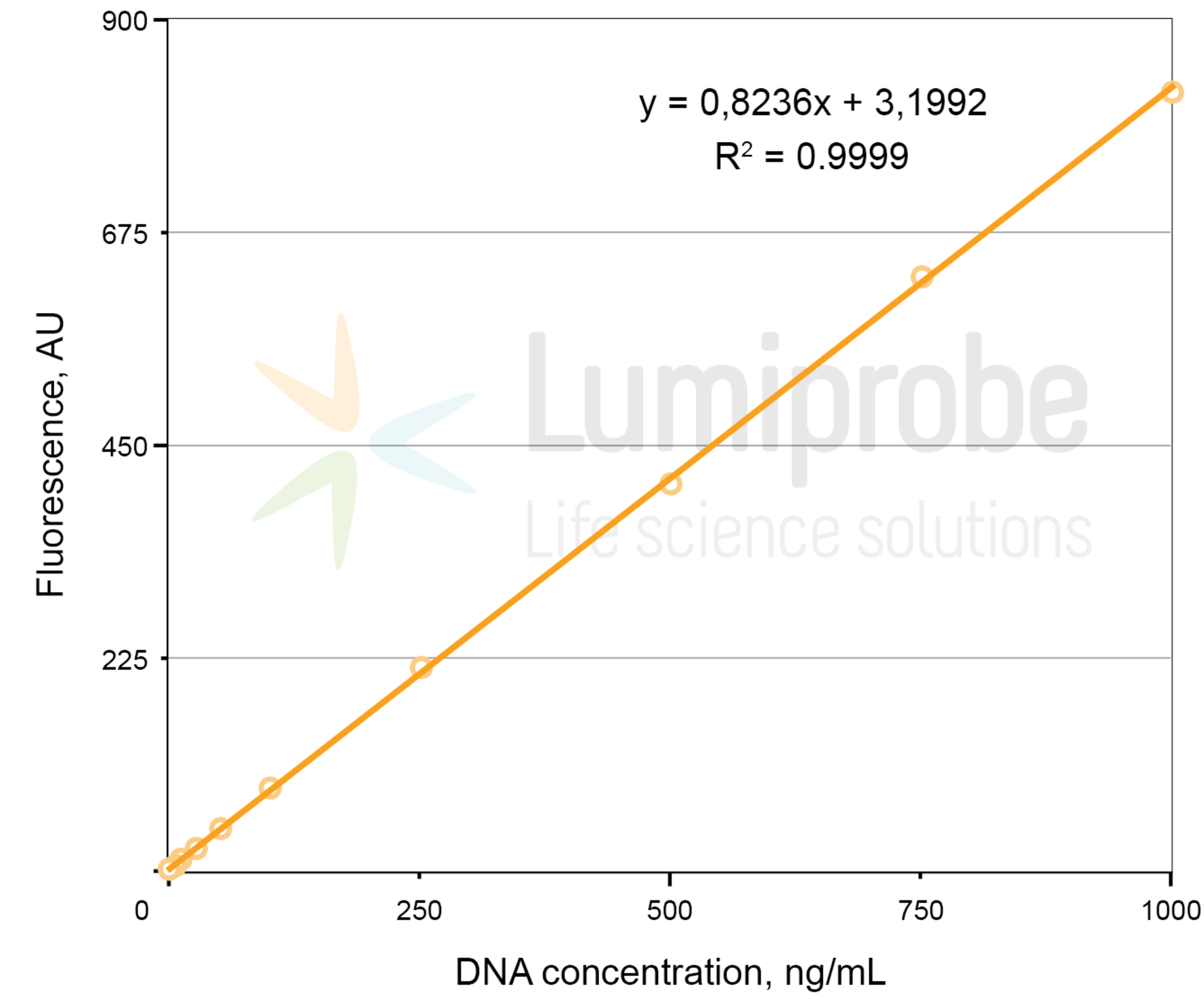
Pico488 测量 DNA 浓度的线性范围为 1 pg/μL — 5 ng/μL。为了实现精确、可重复的荧光测量,我们建议在 TE 缓冲液(10 mM Tris-HCl,pH 7.5,1 mM EDTA)中稀释 DNA 和 Pico488。为了计算 DNA 浓度,我们建议首先使用一系列 DNA 标准稀释液建立校准曲线。
Lumprobe 销售各种包装的Pico488 DNA 定量溶液和Pico488 DNA 定量试剂盒。除了 Pico488 溶液之外,该试剂盒还包括缓冲液和 DNA 标准储备溶液。如果测定体积等于 2 ml,则试剂盒提供的染料溶液量足以分析标准荧光比色皿(体积 3.5 ml)中的 200 个实验数据点。如果使用其他类型的设备来检测荧光,则可以重新调整测定范围。下表提供了常用荧光设备的推荐检测体积。
协议
1. Pico488工作液的制备
解冻 Pico488 染料瓶中的内容物,充分混合,并用缓冲液将所需量的 Pico488 染料溶液稀释 200 倍(我们建议使用 10 mM Тris HCl、1 mM EDTA,pH 7.5)。混合并在3小时内使用。每个实验数据点的 Pico488 工作溶液体积应等于测定体积的 50%(查看下表以查找适合您的荧光测定设备类型的推荐体积)。为所有实验数据点(针对您计划分析的所有样品和所有 DNA 标准稀释液)准备足够的 Pico488 工作溶液。还包括额外 10-25% 的 Pico488 工作溶液体积,以排除可能的移液错误。要计算稀释后的 Pico448 的体积,可以使用以下公式:
V Pico488 = 5/8 × V测定× (N 个样品+ N 个标准品), 其中 V测定是样品或标准品的测定体积,mL,N 个样品 是您计划分析的样品数量,N 个标准品是您计划分析的标准品数量(包括空白样品)。
2. 样品溶液的制备
在缓冲液中稀释 DNA 样本,使溶液体积等于测定体积的 50%(您可以使用任意量的 DNA)。添加等体积的 Pico488 工作溶液。混合并孵育 5 分钟。同样,制备 DNA 标准品的稀释液。请注意,DNA 标准品的稀释度应在样品中 DNA 浓度的范围内。 DNA 标准储备液仅随Pico488 DNA 定量试剂盒提供。Pico488 DNA 定量解决方案的用户 应使用自己的 DNA 标准品。
使用Pico488 DNA 定量溶液进行 DNA 定量的推荐体积:
| 设备类型 |
测定体积 |
稀释后的 Pico488 体积 |
稀释 DNA 体积 |
| 标准荧光比色皿(3.5 ml) |
2毫升 |
1毫升 |
1毫升 |
| 其他荧光比色皿 |
约比色皿体积的 75% |
比色皿体积的 37.5% |
比色皿体积的 37.5% |
| 96 孔板*,每孔 |
0.2毫升 |
0.1毫升 |
0.1毫升 |
| 24 孔板,每孔 |
1毫升 |
0.5毫升 |
0.5毫升 |
| 其他板材 |
约占井容积的75% |
井体积的37.5% |
井体积的37.5% |
| NanoDrop™ 3300* |
0.1毫升 |
0.05毫升 |
0.05毫升 |
* 为了保持测量的准确性和精密度,我们建议避免移液量低于 2 µL。
3. 荧光测量
使用适当的吸收和发射波长或滤光片测量标准和样品 DNA 溶液的荧光(双链 DNA 结合的 Pico488 染料吸收最大波长为 503 nm 的光,发射最大波长为 525 nm 的光)。
4. DNA浓度的计算
绘制荧光与 浓度的关系图,并在任何软件中应用线性回归函数,以获得反映荧光 ( FL ) 与浓度 ( C ) 依赖性的线性方程:
FL = A × C + B。
要计算稀释样品中的 DNA 浓度,请使用以下公式:
C样品= (FL样品 — B)/A,其中FL样品是样品溶液荧光。
要计算未稀释样品中的 DNA 浓度,请使用以下公式:
С init = V Assay × C Sample / V init,其中 V Assay是测定体积(mL),V init是初始 DNA 样品的体积(μL)
或者,您可以使用我们的dsDNA 定量和稀释 计算器完成所有必要的计算。
线性回归示例:荧光与 DNA 浓度